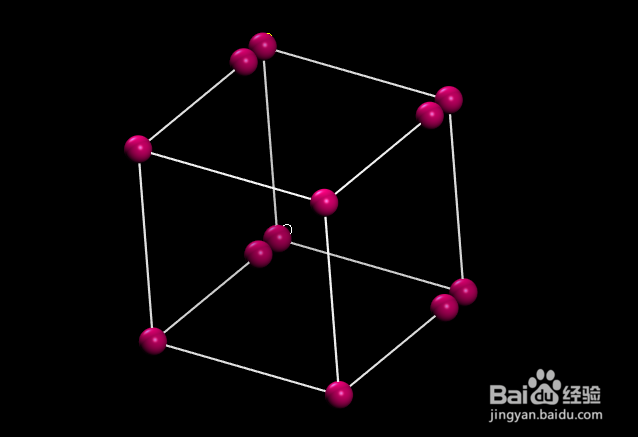
如何用VASP计算分子的键能
1、POSCAR文件为
O dimer in a box 1.0 ! universal scaling parameters
8.0 0.0 0.0 ! lattice vector a(1)
0.0 8.0 0.0 ! lattice vector a(2)
0.0 0.0 8.0 ! lattice vector a(3)
2 ! number of atoms
cart ! positions in cartesian coordinates
0 0 0 ! first atom
0 0 1.22 ! second atom
将氧分子放在一个立方体中

2、INCAR文件为
SYSTEM = O2 dimer in a box
ISMEAR = 0 ! Gaussian smearing
ISPIN = 2 ! spin polarized calculation
NSW = 5 ! 5 ionic steps
IBRION = 2 ! use the conjugate gradient algorithm(使用共轭梯度算法)

3、KPOINTS文件为
Gamma-point only
0
Monkhorst Pack
1 1 1
0 0 0

4、我们应该在INCAR文件中选择几何优化。本例中最多算5个离子步。优化中使用了共轭梯度(CG)算法(IBRION=2)。CG算法需要沿着搜索方向最小化优化。这是布伦特算法的一种变体。
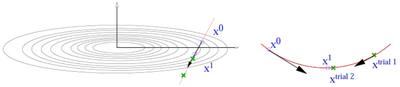
5、计算过程如下,其中
quantitiy trial-energy change是试验步骤中能量的改变

6、实验步数可以由POTIM参数控制,优化后的模型会存于CONTCAR中,如果要继续优化,可以将CONTCAR复制为POSCAR。